

Diagnosis & Management of MSMDS
Diagnosing MSMDS: a medical challenge
Multisystemic Smooth Muscle Dysfunction Syndrome (MSMDS) is an ultra-rare genetic disease difficult to recognize at first glance. Its symptoms appear in different organs and are often confused with more common diseases, delaying diagnosis.
Early diagnosis can save lives and change the course of the disease.
Important note: The information on this page is not a substitute for medical advice. Always discuss it with your healthcare team.
Understanding the diagnostic challenge
What makes MSMDS particularly challenging to diagnose is that its manifestations can appear at different moments in life and vary in severity. Some signs are present from birth, while others develop progressively over time.
For this reason, diagnosis depends not only on identifying individual symptoms, but on recognizing how they connect.
When multiple findings affect organs that rely on smooth muscle function (such as blood vessels, lungs, digestive system, or bladder) MSMDS should be considered.
Early recognition of this pattern is essential to guide genetic testing, avoid unnecessary risks, and ensure appropriate medical care.
When to think about MSMDS
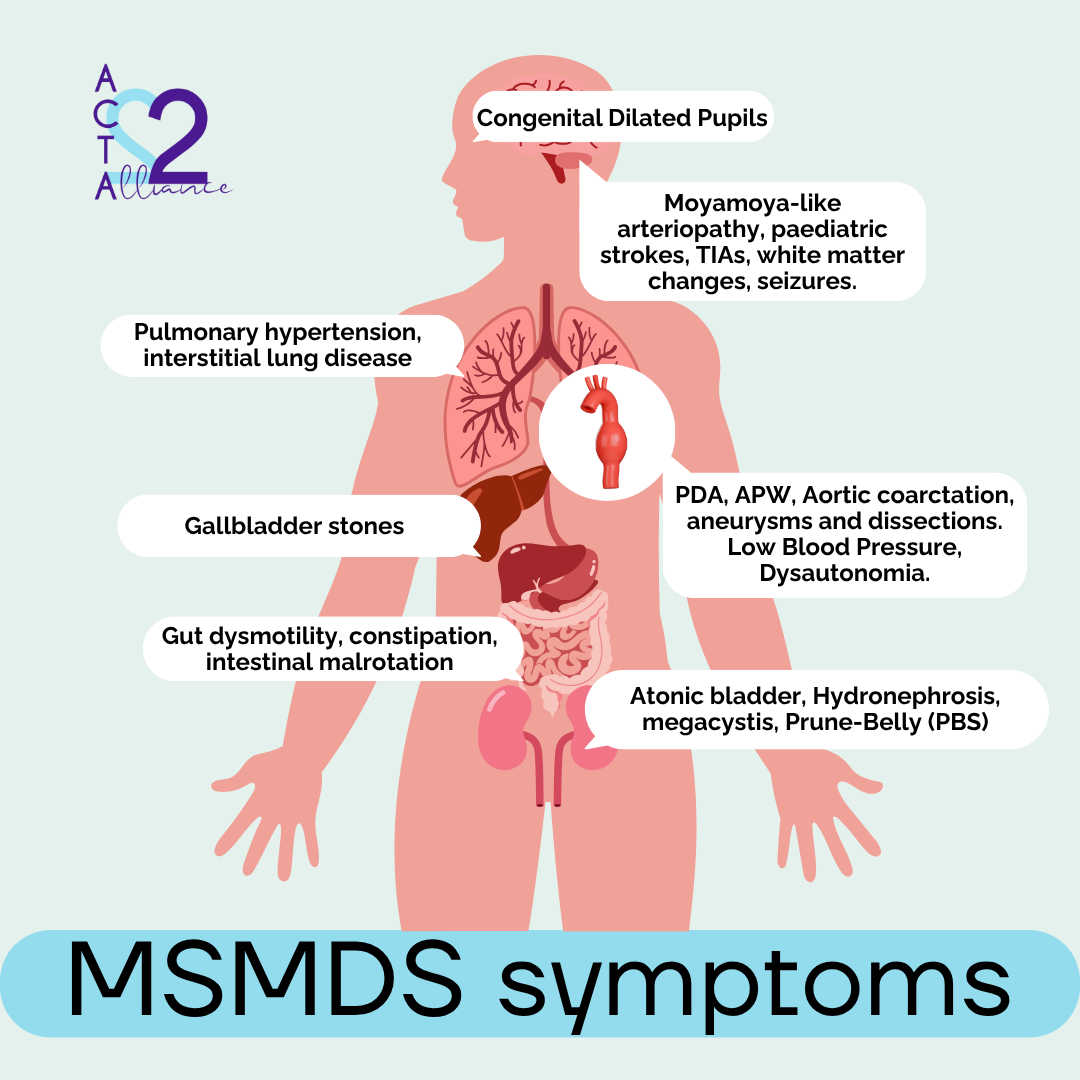
Certain combinations of signs should raise suspicion, especially when they appear early in life.
Early diagnosis of MSMDS may be facilitated when two or more of these features coincide:
- Congenital mydriasis without response to light
- Congenital heart disease such as patent ductus arteriosus (PDA) or aortopulmonary window (APW)
- Cerebral white matter abnormalities in newborns
- Aortic aneurysms in children or adolescents
- Out of the blue strokes in children with a history of cardiac surgery for PDA or APW
- Multisystem involvement (digestive, urinary, respiratory) without a clear explanation
Highlight: If two or more of these signs are present, MSMDS should be considered and a genetic evaluation requested.
Diseases with which MSMDS is confused
Due to its extreme rarity and the wide range of symptoms, MSMDS is often confused with other partial diagnoses. While some of these may be correct in the short term, they do not explain the entirety of the clinical picture or the connection between the complications that arise over time. Among the most common are:
- Moyamoya:
Childhood TIA’s, strokes and cerebral arteriopathy can confound the diagnosis, but MSMDS is differentiated by the absence of basal collateral vessels and by the presence of abnormally straight cerebral vessels.
- Aniridia:
Congenital mydriasis can be interpreted as the absence of an iris. In reality, the iris is present but does not respond to light, causing the common photophobia in children with MSMDS.
- Asthma: Respiratory difficulties are mistaken for childhood asthma, delaying the identification of a generalized smooth muscle dysfunction, which leads to complications such as tracheomalacia, undeveloped alveoli, and interstitial lung disease (ILD) .
These partial diagnoses not only increase families' uncertainty but also delay referral to specialists familiar with MSMDS. Consequently, a lack of adequate information can put the lives of the patients at risk.
A critical example is surgical interventions or tests such as MRI: in children with MSMDS, Anesthesia or sedation with standard protocols can trigger serious complications, including strokes.
To minimize permanent damage, it is essential that medical teams understand the specifics of this disease before any procedure.
How is MSMDS confirmed today?
The most accurate way to confirm MSMDS is through genetic testing, usually performed on a simple saliva sample. These tests analyze genes involved in smooth muscle and connective tissue function like ACTA2 and MYH11.
Doctors and researchers recommend genetic testing when two or more key symptoms appear together, most commonly congenital mydriasis (fixed dilated pupils) and patent ductus arteriosus.
Before referral to a genetics specialist, clinicians may perform:
- Physical examination: to assess the pupillary reflex and rule out similar conditions such as Marfan syndrome.
- Diagnostic imaging: including heart or aortic ultrasound (if the ductus has not been evaluated) and retinal examination to look for vascular tortuosity.
Because different mutations may lead to MSMDS, identifying the exact genetic variant is essential to guide monitoring and care.
Highlight: If you suspect MSMDS, ask your doctor for a genetic evaluation.
What happens after receiving an MSMDS diagnosis?
The options vary depending on the hospital and the specific case, but the first step would be to schedule a series of appointments with neurology, cardiology, pulmonology, gastroenterology and any other necessary specialties. Each department will carry out specific tests to establish the basis of the patient's clinical picture such as:
- MRI with MRA-TOF in brain and blood vessels of the head and neck to evaluate the current state of disease burden and monitor disease progression. Preferred over computed tomography (CT)/CT angiography (CTA) due to higher image resolution for evaluating parenchymal involvement.
- Genitourinary ultrasound as an initial workup.
- Pulmonary function test (PFT) to measure lung efficiency and determine the degree of involvement of smooth muscle cells.
- Images of the vasculature of the chest, abdomen, and pelvis are recommended after diagnosis and every 12 months from age 10.
- Neuropsychological assessments especially in cases with learning or developmental difficulties or when the MRI shows abnormalities of the white matter or stroke.
Depending on the patient's symptoms, some of these tests may be unnecessary, while others will be required. Each case is unique as well as the management and medical care of each person affected with Multisystemic Smooth Muscle Dysfunction Syndrome.
Why is MSMDS so difficult to diagnose?
The path to an accurate diagnosis is often long and complex. The main challenges:
- Extremely low prevalence (<20 cases per million).
- Symptoms affecting multiple systems that appear unrelated
- Limited awareness among healthcare professionals
- Overlap with more common conditions
- Lack of standardized medical protocols
As a result, many families experience a prolonged and difficult journey before receiving an accurate diagnosis.
Why Early Diagnosis Matters
Reaching a diagnosis of MSMDS is not just about naming the disease. It’s about preventing life‑changing risks that can escalate in a matter of hours.
An early diagnosis means more time, fewer complications, and access to the right care from the very beginning.
Because with MSMDS, the challenge isn’t only diagnosing.
It’s diagnosing in time.
Our Commitment to Early Diagnosis
At ACTA2 Alliance, we know that early diagnosis can change the entire future of a person with MSMDS. To make that possible, we focus on:
- Promote awareness of MSMDS in differential diagnosis
- Develop early‑detection protocols, such as pupillary reflex checks in newborns with ductus arteriosus or pediatric stroke.
- Share clinical knowledge with healthcare professionals worldwide.
- Support medical training in ultra‑rare smooth muscle and connective tissue diseases.
- Provide multilingual resources to help identify undiagnosed cases globally.
Early diagnosis saves lives. Every child identified before a major complication gains time, treatment options, and a better chance of avoiding irreversible outcomes.
Literature
1. Zhou YL, Zhang YY, Cheng BL, Xu D, Tang LF, Chen ZM. Multisystemic smooth muscle dysfunction syndrome in children: a case report and literature review. Chinese Journal of Pediatrics, 2017.
https://europepmc.org/article/med/28822439
2. Nadine McCrea, Heather J. Fullerton, and Vijeya Ganesan.
Genetic and Environmental Associations With Pediatric Cerebral Arteriopathy: Insights Into Disease Mechanisms. Stroke, 2019
https://www.ahajournals.org/doi/10.1161/STROKEAHA.118.020479
3. Mc Glacken-Byrne, A.B., Prentice, D., Roshandel, D. et al. High-resolution iris and retinal imaging in multisystemic smooth muscle dysfunction syndrome due to a novel Asn117Lys substitution in ACTA2: a case report. BMC Ophthalmol, 2020.
https://bmcophthalmol.biomedcentral.com/articles/10.1186/s12886-020-01344-w#citeas
4.
Interstitial Lung Disease in MSMDS: Developmental Smooth Muscle Dysfunction in the Lungs. Speaker: Dr. Bernard Kinane (MGH). Presented at the 2025 MSMDS Conference.
https://www.youtube.com/watch?v=dVMH-glRu90&list=PL9i5mdmWtAp4VhC76CV1OJ2WJNtnNXME7&index=15
5. Nicholas Houska, Michal Schafer, Kathryn C. Chatfield, Timothy J. Bernard, Richard J. Ing.
Anesthetic Considerations for Children With Multisystem Smooth Muscle Dysfunction Syndrome and Review of the Literature, Journal of Cardiothoracic and Vascular Anesthesia, 2022.
https://www.sciencedirect.com/science/article/abs/pii/S1053077022002786
6. Chen SN, Wang YQ, Hao CL, Lu YH, Jiang WJ, Gao CY, Wu M.Multisystem smooth muscle dysfunction syndrome in a Chinese girl: A case report and review of the literature. World J Clin Cases, 2019.
https://www.wjgnet.com/2307-8960/full/v7/i24/4355.htm
7.
Latest clinical recommendations on MSMDS imaging, Dr. Diana Tambala, MD (MGH). Presented at the 2025 MSMDS Conference. 2025.
https://youtu.be/Gz-sHg-kFRQ?si=gJGK9KUb_Wv5H306&t=1851
8. Petrov I, Keltchev A, Stankov Z, Vasilev S.The journey of a patient with ACTA2 mutation – literature review and case report.Cor Vasa 2022;64:619–621.https://e-coretvasa.cz/pdfs/cor/2022/06/08.pdf
